JOURNAL 1297
Organic Communications
Year: 2019 Issue: 2 April-June
p.115 - 119
Viewed 2817 times.
GRAPHICAL ABSTRACT
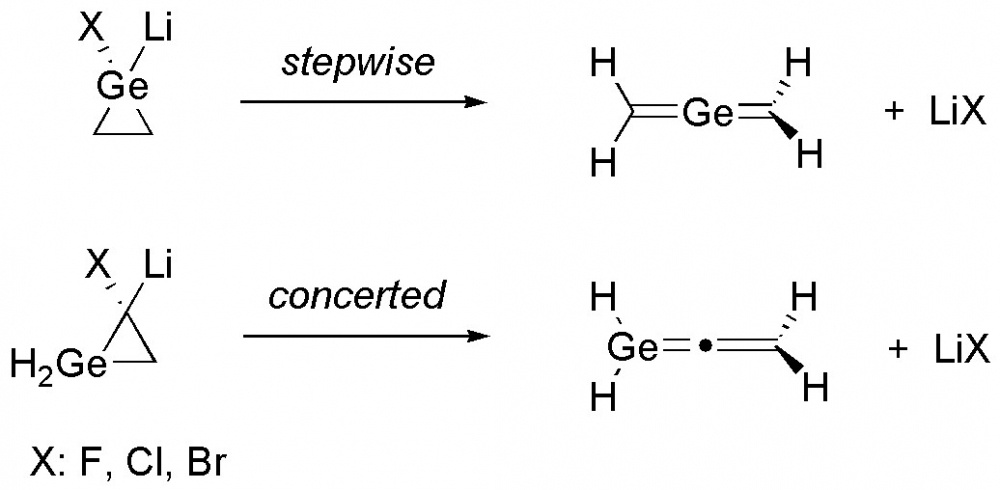
ABSTRACT
Density functional theory calculations were employed to explore the ring–opening reaction mechanisms of mono germanium analogues of cyclopropylidenoids via Doering–Moore–Skattebøl method. The theoretical findings revealed that the stepwise fashion with the intermediacy of a free germacyclopropylidene is operative for the structure with germanium atom on the carbenic position (1). Moreover, the cyclopropylidene analogue (4) of the title structures proceeds via concerted manner for the corresponding ring-opening reaction. The calculated overall energy barrier to trigger the stepwise ring-opening reaction for 1 is strongly higher than the concerted fashion for 4. Additionally, the effect of halogens (X = F, Cl, Br) on the reaction mechanism were also investigated. The energy barriers for chlorine substituted precursors are found to be lower than the fluorine and bromine substituted forms.
KEYWORDS- Germaallene
- reaction mechanism
- DFT
- germanoid
- germylene
